此次備置測試的樣本取得和標準:(寄給黃雅怡 博士的信件內容)
Test Run會以HiSeq 2500 pairend 100 bp定序,使用限制酶為PstI。這個lane中會有約40~50個個體間,所以會需要你們的個體數約有十個。(另外我們還有其他預計樣本:構樹*30、龍膽屬*10)由於實驗要一起執行,但因其他樣本都尚未備齊,預期在八月中下旬開始製備。所需樣本DNA條件如下:
1. 1μg genomic DNA,濃度 >25 ng/μL
也就是1μg DNA要溶於小於44 μL的水,不符合的話,請濃縮,或DNA量要足夠。DNA量"至少 “1 μg,如果可以多一點會更好。
2. 回溶DNA的液體請用水,不要TE buffer
特別注意絕對不要有鹽份和酒精的殘留,在抽取DNA時酒精要充分揮發。
3. DNA品質,不可有二次代謝物或色素
我是使用Monarch® PCR & DNA Cleanup Kit (5 μg)。
請記得所第1.點要求的DNA濃度是在純化後的濃度,簡單說DNA的質和量都要達標準。
4. DNA片段大小完整,不要smear
此次測試注意:
1. 不要來源難以取得或DNA量稀少的樣本
我過去是在台大別人的研究室做這個實驗,故目前尚未在中研院研究室獨立做過RAD-Seq Library製備,所以此次test run主要目的是以測試、建立經驗為主。等此次後成功之後,再來嘗試那些樣本比較保險。(簡單說,若不小心失敗,我會自責毀了你們的心血)
2. 預計在8/13那禮拜給我樣本
如果可以,先寄給我看一下DNA的相關品質資料:260/280 ratio、260/230 ratio(nanodrop)、ng/μL(Qubit)和膠圖。
___________________________________
9/17 使用Sample如下:
P1 Adapter
|
|
|
ng/uL
|
需要1ug所需的量 (uL)(1ug=1000ng)
|
實驗用量(四捨五入)
|
補水(總體積44uL)
|
1
|
CF023
|
KFC 3604-4
|
93
|
10.75268817
|
11
|
33
|
2
|
CF025
|
KFC 3604-6
|
99.8
|
10.02004008
|
11
|
33
|
3
|
CF026
|
KFC 3605-3
|
92.4
|
10.82251082
|
11
|
33
|
4
|
CF027
|
KFC 3605-4
|
106
|
9.433962264
|
10
|
34
|
5
|
CF008
|
KFC 3606-1
|
45.2
|
22.12389381
|
23
|
21
|
6
|
CF028
|
KFC 3606-3
|
110
|
9.090909091
|
10
|
34
|
7
|
CF029
|
KFC 3608-3
|
102
|
9.803921569
|
10
|
34
|
8
|
CF030
|
KFC 3608-4
|
64.6
|
15.47987616
|
16
|
28
|
9
|
CF012
|
KFC 3609
|
28.4
|
35.21126761
|
36
|
8
|
10
|
CF013
|
KFC 3610
|
36.8
|
27.17391304
|
28
|
16
|
11
|
CF034
|
KFC 3611
|
70.6
|
14.16430595
|
15
|
29
|
12
|
CF037
|
KFC 3613-5
|
98.8
|
10.12145749
|
11
|
33
|
13
|
CF038
|
KFC 3614-1
|
66.4
|
15.06024096
|
16
|
28
|
14
|
CF016
|
KFC 3615
|
33.4
|
29.94011976
|
30
|
14
|
15
|
CF018
|
KFC 3616-2
|
45
|
22.22222222
|
23
|
21
|
16
|
CF042
|
KFC 3617-2
|
31.4
|
31.84713376
|
32
|
12
|
17
|
CF043
|
KFC 3618-1
|
29
|
34.48275862
|
35
|
9
|
18
|
CF045
|
KFC 3618-3
|
76
|
13.15789474
|
14
|
30
|
19
|
CF046
|
KFC 3619-1
|
104
|
9.615384615
|
10
|
34
|
20
|
CF047
|
KFC 3619-2
|
52.6
|
19.01140684
|
20
|
24
|
21
|
CF051
|
KFC 3619-6
|
90.6
|
11.03752759
|
12
|
32
|
22
|
CF058
|
KFC 3619-G
|
94.2
|
10.61571125
|
11
|
33
|
23
|
CF060
|
KFC 3620-2
|
69.4
|
14.4092219
|
15
|
29
|
24
|
CF061
|
KFC 3620-3
|
54.4
|
18.38235294
|
19
|
25
|
25
|
CF062
|
KFC 3621
|
27.8
|
35.97122302
|
36
|
8
|
26
|
CF067
|
KFC 3625
|
31.2
|
32.05128205
|
33
|
11
|
27
|
CF068
|
KFC 3628
|
47.4
|
21.09704641
|
22
|
22
|
28
|
CF069
|
KFC 3629
|
38.2
|
26.17801047
|
27
|
17
|
29
|
C252
|
KFC3601_6
|
156
|
6.41025641
|
7
|
37
|
30
|
CF073
|
KFC 3643-4
|
108
|
9.259259259
|
10
|
34
|
31
|
|
FG26
|
37
|
27.02702703
|
28
|
16
|
32
|
|
GB1
|
63.8
|
15.67398119
|
16
|
28
|
33
|
|
GB2
|
39.8
|
25.12562814
|
26
|
18
|
34
|
|
LM15
|
39.4
|
25.38071066
|
26
|
18
|
35
|
|
LM19
|
51.6
|
19.37984496
|
20
|
24
|
36
|
|
LM3
|
71.4
|
14.00560224
|
15
|
29
|
37
|
|
MD7
|
38.2
|
26.17801047
|
27
|
17
|
38
|
|
SUGM19
|
63.2
|
15.82278481
|
16
|
28
|
39
|
|
WA2-5
|
39.6
|
25.25252525
|
26
|
18
|
40
|
|
WK7
|
36.2
|
27.62430939
|
28
|
16
|
41
|
|
4
|
28.7
|
34.84320557
|
35
|
9
|
42
|
|
12
|
31.4
|
31.84713376
|
32
|
12
|
43
|
|
20
|
34
|
29.41176471
|
30
|
14
|
44
|
|
23
|
27.1
|
36.900369
|
37
|
7
|
45
|
|
24
|
27
|
37.03703704
|
38
|
6
|
46
|
|
25
|
29.28
|
34.15300546
|
35
|
9
|
47
|
|
27
|
32.9
|
30.39513678
|
31
|
13
|
48
|
|
32
|
25.2
|
39.68253968
|
40
|
4
|
49
|
|
33-2
|
28.4
|
35.21126761
|
36
|
8
|
50
|
|
24-1
|
25.4
|
39.37007874
|
40
|
4
|
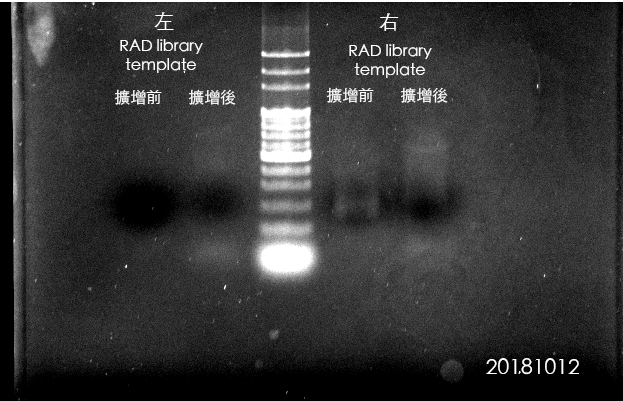
P1 Adapter 1~30為構樹(DNA來自嘉瑢)、31~40為珊瑚(DNA來自黃雅怡博士)、41~50為龍膽(DNA來自景淞)。濃度為DNA抽取者所提供。在實際操作時發現:在做的時候發現黃博士給我的十個sample,有七個sample的DNA量都不足1ug。如FG26濃度為37 ng/ul,那至少要有28 ul,才有1ug (37*28=1036 ng),不過他們的sample的體積都只有約15 ul。由於事先沒有再次檢查,但因為已經做下去又臨時無法改變,所以我只用了其中的三管(GB1、LM3、SUGM19)。
___________________________________
實驗protocol outline:這是由我改寫的protocol,此步驟在我的碩士論文研究中有使用過(當時擴增的效率並沒有使用陳老師protocol的好,故最後定序是使用後者protocol的樣本)。李宜軒亦使用此套protocol,在李宜軒的論文中有詳細記載。
主要使用陳凱儀 老師lab的protocol
1.2 限制酶切割
1.3 P1 Adapter連結酶反應
1.4 樣本合併
1.5 混合樣品DNA分子的隨機斷裂
1.6 DNA的濃縮與片段篩選
後續使用NEBNext® Ultra™ II DNA Library Prep Kit for Illumina®的protocol
1.1 NEBNext End Prep
1.2 Adaptor Ligation
1.3 Size Selection or Cleanup of Adaptor-ligated DNA
1.4 PCR Enrichment of Adaptor Ligated DNA
1.5 Size Selection or Cleanup of Amplification
___________________________________
1.2 限制酶切割(9/18 17:00 – 9/19 8:00)
潛在問題:限制酶 Pst-HF(2016/11過期)為台大所帶過來,是否已失效?
1.3 P1 Adapter連結酶反應(9/19)
1.4 樣本合併(9/19、9/20)
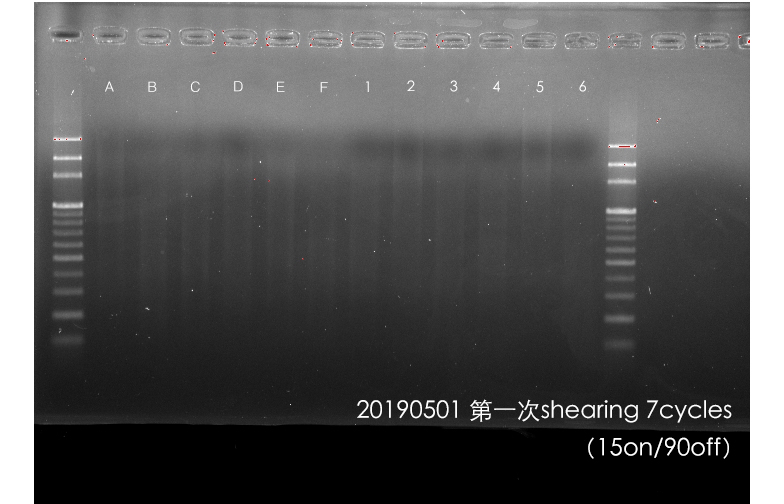
此步驟有個主要變因為DNA破碎機器改變:為使用Biouptor Pico。劉世慧 博士的經驗為:1. 大於3K的DNA經過三個循環(一循環為一次啟動/停止,皆為30秒),即可達300~700 bp。2. 裝取100ul的效率比裝載50ul的破碎效率好。故當時將陳老師的Protocol(每一個離心管裝50ul,至少總共21個循環)調整成:每一個離心管裝100ul,初次以2個循環,跑膠檢視片段大小,在逐一增加循環次數。
9/19:調整:每一管裝載100ul,故每個接完P1的樣本需各取28ul,最後合併為1200ul樣本,再分裝至12管0.65ml的microtube中。(注意!調整成100ul,預期外的問題*出在後續 1.6 DNA的濃縮與片段篩選。)
9/20:未調整:每一管裝載50ul,故每個接完P1的樣本需各取15ul,最後合併為600ul樣本,再分裝至12管0.65ml的microtube中。連結完成P1 Adapter的每個樣品體積為60ul,經9/19、9/20 兩次使用,目前剩餘60-28-15=17ul(可從此步後續從新開始)。
1.5 混合樣品DNA分子的隨機斷裂
9/19:每一管裝載100ul,累加循環2、2+4、2+4+10、2+4+10+10循環。此測試發現:並沒有發生劉博經驗中的高破碎效率(差異可能為DNA的溶液差別?)。最後試出來的結果,和陳老師Protocol的次數相去不大。故我將此步驟維持為20個循環。
這個slideshow需要JavaScript。
9/20:每一管裝載50ul,直接10+10循環。此測試發現:大部分的片段為200~500bp左右。(9/20接下來僅用前面6管,剩餘六管可從此步驟開始。)(*20181017更新:此六管剩餘已於20181017 RAD-Seq Library備置測試使用完畢。)

1.6 DNA的濃縮與片段篩選
9/19、9/20 濃縮:此步驟的DNA濃縮,使用Monarch® PCR & DNA Cleanup Kit (5 μg) 四管回收,Wash buffer步驟僅使用一次(Kit protocol中為兩次),使用水回溶,最後合併約70ul的溶液。
9/19 片段篩選:在DNA破碎的步驟,每一管裝載100ul,故DNA量其實為陳老師protocol量的兩倍,此時發現的預期外問題*為,DNA量增加兩倍,導致AMPure磁珠在操作上有很大的問題:幾乎無法受到磁鐵的吸引,磁珠和溶液無法分離。(9/19這管的實驗至此步停止)
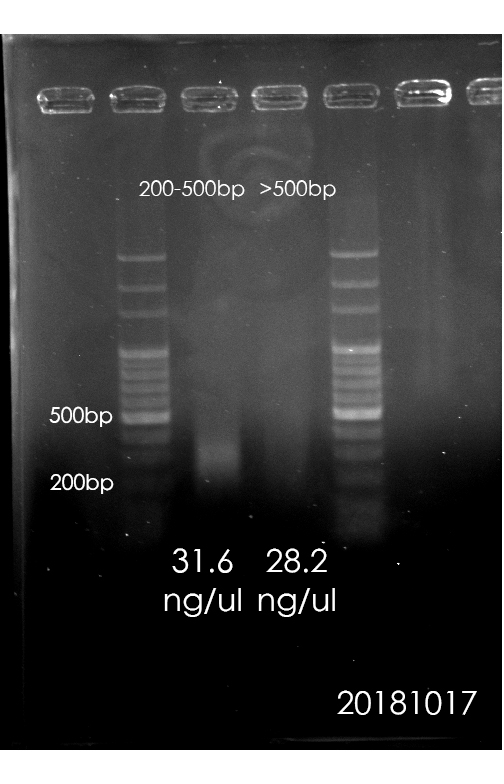
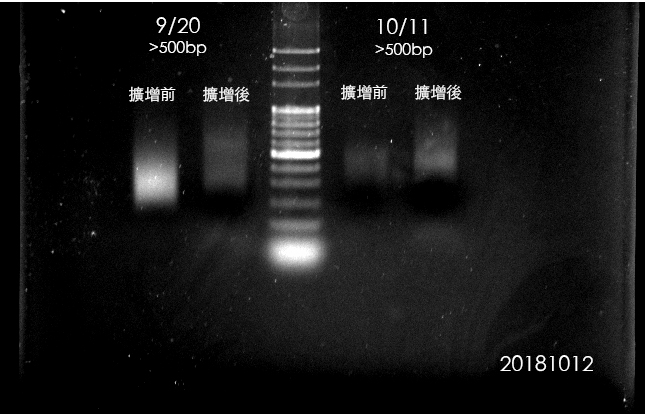
9/20 片段篩選:在DNA破碎的步驟,每一管裝載50ul,在AMPure操作上順利。(接下來步驟由9/20這管繼續)階段目標的膠圖和濃度:片段為200~500bp之間,濃度為94.4ng/ul,回溶23ul(-2ul於跑膠和測濃度)。取DNA 1ug(此次使用為10ul,還剩餘約10ul,還可從此步後續從新開始)(*20181012更新:此管剩餘已於20181004-12 RAD-Seq Library備置測試使用完畢。)繼續下一步驟。

後續使用NEBNext® Ultra™ II DNA Library Prep Kit for Illumina®的protocol
1.1 NEBNext End Prep
使用9/20這管,10ul(濃度94.4ng/ul)開始此步驟。
1.2 Adaptor Ligation
我這次測試P2 Adapter 10uM(自製)使用量:2.5ul。(宜軒:10uM, 1ul、Kit官方Adapter:15uM, 2.5ul)潛在問題:是否P2 Adapter放太多了?
1.3 Size Selection or Cleanup of Adaptor-ligated DNA
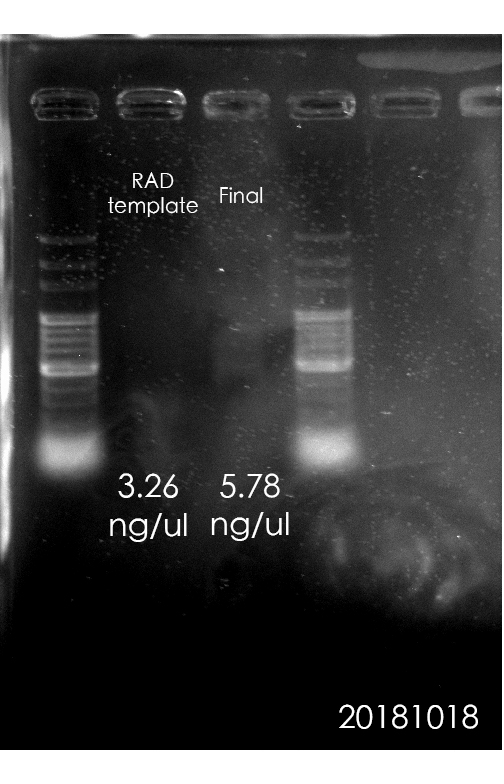
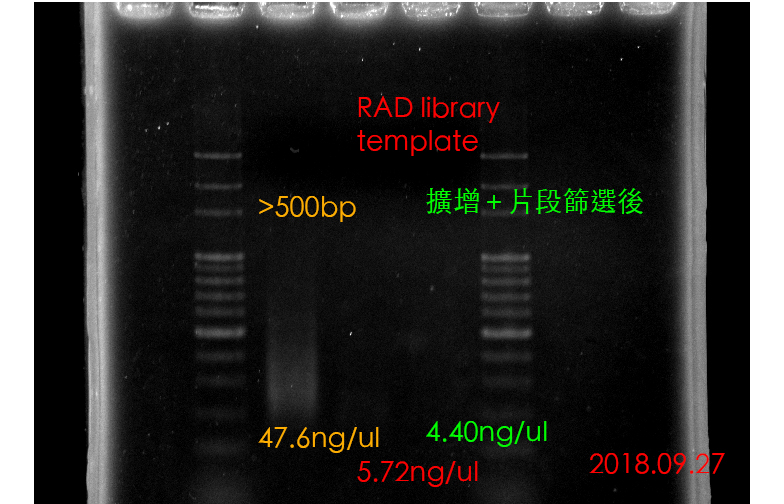
片段篩選後的RAD library template濃度為:5.72ng/ul(protocol階段目標至少為:4ng/ul,最後體積為15ul,DNA量才能達50ng。)
1.4 PCR Enrichment of Adaptor Ligated DNA
自製Solexa primer 10uM各放5ul,並使用Kit protocol中的PCR擴增程式。潛在問題:Solexa primer為台大所帶過來,為去年(2017)購買,是否失效?
1.5 Size Selection or Cleanup of Amplification
使用陳老師protocol的AMPure的片段篩選。結果並未擴增成功。潛在問題:沒有測試到單純擴增後的濃度和膠圖,不知是否為最後一步片段篩選的問題,或是真的擴增失敗。
___________________________________
潛在問題 總整理:
- 限制酶 Pst-HF為台大所帶過來,為去年(2017)購買,是否失效?
- 是否P2 Adapter放太多了?
- Solexa primer為台大所帶過來,為去年(2017)購買,是否失效?
- 擴增失敗原因,不知道是最後一步片段篩選的問題,或是真的擴增失敗?(擴增失敗原因可能為?)
___________________________________
20181002 妤馨 建議:
我看了你的實驗紀錄。對於enzyme的活性有些懷疑。因為你是做complete digestion,可是在電泳圖看來,還有很多intact DNA。這代表這能有很多DNA沒有被切斷。不過這也有可能是酵素的特性。
不知道你之前使用這個酵素的經驗,是否也有很多intact DNA存留的情況
我以前測試酵素時,會拿一些DNA切切看,然後將具有相同濃度有切跟沒有切的DNA一起跑電泳,然後就會知道酵素有沒有切動以及切的效率為何
不知道你或宜軒以前成功的library是否有留下?假如你想知道Solexa primer是否失效,可以把以前已知濃度的library當作template,然後PCR看有沒有amplify成功
還有,實驗室的磁珠已經過期,雖然學姊有測試過沒有問題,還是得小心使用。
在1.6電泳圖裡,長度好像跟你標示的都不一樣,大部份的片段都集中在150bp? 有點不確定這個marker長度
在10+10的電泳圖裡,ladder最下面那一團不是100bp吧?假如我沒搞錯,這樣蠻多管的長度都只有100-200bp耶…
我 回覆:
1. 酵素反應後的片段大小。這點我覺得是我忽略的部分。我們之前不會特別測試酵素的活性(像是看膠圖這類的),這點我覺得很重要,非常感謝提醒。畢竟酵素也是前年買的,又從台大帶過來,我想可能會有問題。
2. 這次震碎的片段大小都偏小。這是問題。圖上面標示的,是那次膠圖的目標大小,非實際大小。所以這次在DNA破碎上沒有掌控好。因為第一次分次2+4+10+10的流程,最後好像還是有很大片段,所以第二次我就直接連續的10+10,但就震太小了。這我有想過,這之後會在注意。只是覺得這應該不是影響沒有擴增的主因。不過很謝謝提醒。
現在我打算:買新的酵素。感覺舊的酵素(還沒有分裝)比新訂的primert出問題的機率高太多。